-
WHAT WE DO
- CLINICAL SOLUTIONS
- SERVICES
-
WHO WE ARE
-
RESOURCES

CDISC standards have become an integral part of the life science industry; nevertheless, we will have to continue to deal with clinical data in different legacy formats for some time in the future. While the use of purely CDISC-formatted data from the very beginning of a submission project is unproblematic, combining data in legacy format with CDISC standardized data presents considerable challenges and therefore requires careful planning and special attention.
Scenario 1: “The files you sent are kaput“
XPT files cannot be opened in MS Word. This may seem funny but illustrates a challenge the industry is constantly facing.
Outside the clinical data science world, there is very little understanding of what needs to be done with clinical data for a regulatory submission to the FDA.
Regulatory Affairs departments are hesitant to approach the FDA outside the mandatory milestones. But with legacy data, it is important to contact the agencies with a sound data concept early to leave enough time for data preparation.
The pre-NDA / BLA meetings are usually too late for this discussion and should focus on important science aspects rather than data structures. Requests for the “full CDISC package” with a clean CDISC-Validator log often lead to some unnecessary effort.
Scenario 2: Analysis Datasets ≠ XPT Compliant
Starting Position – Data from multiple studies was analyzed using legacy formats. Dataset and variable names were too long for direct conversion to XPT format.
Possible Solution – Dataset and variable names need to be carefully renamed and standardized across all studies. Programs should be generated and submitted to map data back and forth between the data structures. Old and new names need to be documented in the DEFINE document.
Scenario 3: Comparing Original Results against Mapped Data Project Outline
For many projects only legacy raw data, legacy analysis data and original analysis results are available. Data preparation, analysis programs, and data definition documentation are missing. The customer needs a re-mapping of the legacy raw data to SDTM followed by the creation of CDISC compliant ADaM datasets. As a final QC step analysis results need to be recreated based on ADaM datasets and compared to original analysis results.
QC Result – We often see that discrepancies between the original and the re-programmed analysis emerge. Because of the lack of additional information on the original analysis, the resolution and documentation of findings is extremely time-consuming.
Potential Issues:
Scenario 4: Documentation
More often than not empty folders find their way into the folder tree. Sufficient documentation is key for reviewers to understand where the data came from and how it was processed.
Annotated CRFs and DEFINE documents are needed not only for SDTM data but also for legacy data.
Do not overload single documents. If more information is needed to understand certain aspects of the data, e.g. derivation of the key efficacy parameter, provide documents in addition to the reviewer’s guide and the define document, KISS — keep it short and simple and easy to understand
To Conclude
Every submission project is unique and needs careful planning to avoid costly delays
About MaxisIT
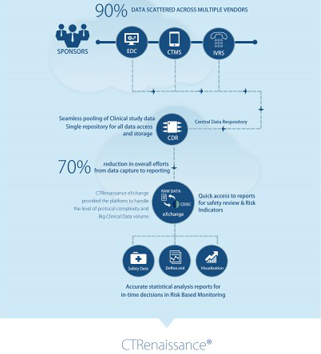
At MaxisIT, we clearly understand strategic priorities within clinical R&D, and we can resonate that well with our similar experiences of implementing solutions for improving Clinical Development Portfolio via an integrated platform-based approach; which delivers timely access to study specific as well as standardized and aggregated clinical trial operations as well as patient data, allows efficient trial oversight via remote monitoring, statistically assessed controls, data quality management, clinical reviews, and statistical computing. Moreover, it provides capabilities for planned vs. actual trending, optimization, as well as for fraud detection and risk-based monitoring. MaxisIT’s Integrated Technology Platform is a purpose-built solution, which helps Pharmaceutical & Life sciences industry by “Empowering Business Stakeholders with Integrated Computing, and Self-service Dashboards in the strategically externalized enterprise environment with major focus on the core clinical operations data as well as clinical information assets; which allows improved control over externalized, CROs and partners driven, clinical ecosystem; and enable in-time decision support, continuous monitoring over regulatory compliance, and greater operational efficiency at a measurable rate”.
25 Mar 2021

17 Apr 2019