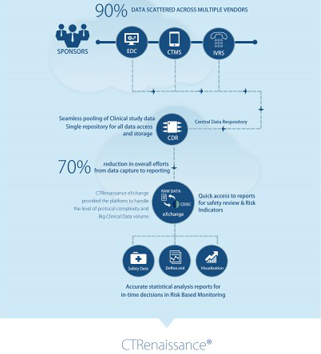
-
WHAT WE DO
- CLINICAL SOLUTIONS
- SERVICES
-
WHO WE ARE
-
RESOURCES
Clinical research is experiencing a revolution with a huge range of connected devices growing in popularity, with wearable and implantable devices across healthcare, fitness tracking and diet. Pharmaceutical companies sponsoring trials are incorporating these devices into ever more elaborate clinical trials, generating ever larger datasets, while sifting through social media streams and their own big data sources. It is now easier than ever before to store, manage and query ever increasing datasets.
The growth in the range of inter-connected devices across healthcare represents an exponential growth in the volume of data collected in ever more elaborate Clinical Trials. This growth in the volume of data presents new challenges for Clinical Data Scientists and requires new solutions and new tools for cross-study data analysis.
To meet these demands, Clinical Data Scientists are increasingly choosing open source solutions to leverage the active open source communities of experienced developers and statisticians. The R scripting language is ever more popular in the biostatistics and statistical programming fields and supports predictive analytics, big data analysis, and offers the potential to leverage Machine Learning and Artificial Intelligence.
Regulators already accept R for statistical analysis and the requirement for skills in R is growing faster than other competing tools. This blog will look at the use of R in cross study data analysis using SDTM data and how and MaxisIT’s SCE can add value to the process.
R – OPEN, EXTENSIBLE, SCALABLE, AVAILABLE
Today, written words and numbers are everywhere, unending and ever – changing. In this world of infinite variety, visuals are still the best way to tell a story. In fact, visualization is more important than ever, because with all the information that’s available, it’s getting harder and harder to sift through the clutter to understand what’s valuable.
Today, visualizations are the best way to filter out the noise and see the signals.
R is a statistical and visual language used by a growing number of data analysts inside corporations and academia, whether being used to set ad prices, find new drugs more quickly or fine-tune financial models. Companies as diverse as Google, Pfizer, Merck, Bank of America and Shell use R.
It is also free. Open-source software is free for anyone to use and modify so statisticians, engineers and data scientists can improve the software’s code or write variations for specific tasks. Packages written for R add advanced algorithms, richly colored and textured graphs and mining techniques to dig deeper into databases. At MaxisIT, we offer an open statistical computing environment that supports R to make it easy for scientists manipulate their own data during nonclinical drug studies rather than send the information off to a statistician.
R IN CROSS STUDY ANALYSIS
Clinical Data Scientists can use pooled data in R from multiple study databases for their visualizations, and train and apply Machine Learning workflows over ever larger datasets. Here is a stepwise process of how R can be used to conduct cross-study analysis.
In summary, R is an excellent tool to connect to the conformed data, and allows sponsors to pool dataframes (datasets) using only a few keystrokes. Sometimes it is a little time consuming to add a bespoke package of our own to allow export to SAS V5 xpt files. MaxisIT offers a Statistical Computing Environment (SCE) that is open and flexibly integrates preferred tools SAS / R applications. The SCE is analytics agnostic & integrated data repository that provides faster access to clinical data.
Key features of MaxisIT’s SCE
REGULATORY CONSIDERATIONS
The FDA’s Statistical Software Clarifying Statement declares that any suitable software can be used in a regulatory submission. Some data-exchange regulations do require the use of the XPT file format, which is an open standard, not restricted to SAS. MaxisIT’s SCE is 21CFR Part 11 and GxP compliant environment offering full traceability, transparency and auditability.
R – THE FUTURE
R use is clearly growing across many industries and it one of the key tools for today’s Clinical Data Scientist. Here is why R is the future of Clinical Development:
Supporting the statistical language R in the most user-friendly manner is MaxisIT’s SCE that delivers ultimate empowerment to Biostatisticians, Clinical Programmers and Data Scientists. It is a completely metadata-driven and scalable Statistical Computing Environment with Integrated Data Repository that supports regulatory analysis without any constraints.
MaxisIT
At MaxisIT, we clearly understand strategic priorities within clinical R&D, and we can resonate that well with our similar experiences of implementing solutions for improving Clinical Development Portfolio via an integrated platform-based approach; which delivers timely access to study specific as well as standardized and aggregated clinical trial operations as well as patient data, allows efficient trial oversight via remote monitoring, statistically assessed controls, data quality management, clinical reviews, and statistical computing. Moreover, it provides capabilities for planned vs. actual trending, optimization, as well as for fraud detection and risk-based monitoring. MaxisIT’s Integrated Technology Platform is a purpose-built solution, which helps Pharmaceutical & Life sciences industry by “Empowering Business Stakeholders with Integrated Computing, and Self-service Dashboards in the strategically externalized enterprise environment with major focus on the core clinical operations data as well as clinical information assets; which allows improved control over externalized, CROs and partners driven, clinical ecosystem; and enable in-time decision support, continuous monitoring over regulatory compliance, and greater operational efficiency at a measurable rate”.
The role of Master Metadata Management (MDM)
MDM is a technology-enabled discipline in which business and IT work together to ensure the uniformity, accuracy, stewardship, semantic consistency and accountability of the enterprise’s official, shared master data assets. The idea of Master Data focuses on providing unobstructed access to a consistent representation of shared information.
How does it work?
Master Data Management (MDM) comprises of a set of processes and tools that consistently define and manage the master data and master reference data of an enterprise, which are fundamental to the company’s business operations. MDM has the objective of providing processes & tools for collecting, aggregating, matching, consolidating, assuring quality, persisting and distributing such data throughout an organization to ensure consistency and control in the ongoing maintenance and application use of this information.
There are different models for master data management – the 2 main extremes are
CHANGING LANDSCAPE: Enforcing data standards from protocol onwards
There are two approaches to enforcing data standards from protocol, they are the retroactive and proactive approaches.
Retro-active approach from paper protocol
Pro-active approach with structural metadata
Efficient Data Integration and compliance with regulatory standards does not start after pooling (retroactive approach); it starts with the protocol (proactive approach)
A proactive approach is based on two components:
To be manageable, variables in an MDR need to be grouped in semantically meaningful “clinical research concepts” (CRC)
To Conclude – Sponsors need to change the way they consider compliance with data standards and data integration: From a retroactive way (building define.xml at submission) to a proactive approach (study data standards defined at study setup)
For that to happen, sponsors need new tools to manage metadata. Such tools should enable
MaxisIT
At MaxisIT, we clearly understand strategic priorities within clinical R&D, and we can resonate that well with our similar experiences of implementing solutions for improving Clinical Development Portfolio via an integrated platform-based approach; which delivers timely access to study specific as well as standardized and aggregated clinical trial operations as well as patient data, allows efficient trial oversight via remote monitoring, statistically assessed controls, data quality management, clinical reviews, and statistical computing. Moreover, it provides capabilities for planned vs. actual trending, optimization, as well as for fraud detection and risk-based monitoring. MaxisIT’s Integrated Technology Platform is a purpose-built solution, which helps Pharmaceutical & Life sciences industry by “Empowering Business Stakeholders with Integrated Computing, and Self-service Dashboards in the strategically externalized enterprise environment with major focus on the core clinical operations data as well as clinical information assets; which allows improved control over externalized, CROs and partners driven, clinical ecosystem; and enable in-time decision support, continuous monitoring over regulatory compliance, and greater operational efficiency at a measurable rate”.
Industry wide Clinical Trial collaborative efforts offers significant improvement over siloed individual databases in providing superior Patient Outcomes. The efforts however were still limited to Rare Disease categories and Data Sources resulting in limited Clinical Analyses and Insight. A Clinical Data Repository utilizing Big Data will enable Pharmaceutical Cos to utilize new Analytic techniques and optimize Patient Journey from Drug Discovery to bedside treatment.
Mining for Big Data enables us to
BIG DATA USE CASES IN PHARMA
Let us consider some possible use cases for Big Data techniques in the domain of Clinical Research.
REPORTING
The more informed a company is on the conduct of its operations, the better placed it is to realize efficiencies. Large amounts of data can become an impediment to good reporting, as the time required to provide results can scale terribly. Big Data techniques make it simpler to generate stratified reports quickly. Using the map function to partition a transactional feed by date make summarizing transactions by hour, day, week, month or quarter simple and using the Lambda Architecture provides the capability for continual real time reporting. Many vendors today offer reporting solution that leverages big data but none as comprehensive as MaxisIT’s Analytics & Reporting platform which satisfies effective decision-making requirements of diverse clinical business functions in a self-services manner. It is completely web-based and scalable platform designed to view large sets of data and provides various simple to advance analytics & reporting functionalities with drag-n-drop configurations for effective reporting.
INTELLIGENT USE OF SOCIAL NETWORKS
Market intelligence can pull from an ever-expanding selection of data sources when looking at the impact of a drug. One of the most valuable is social media, with consumers often referencing a drug or indication. Using tools like the Lambda Architecture we can mine the large feeds of information to pull out interesting messages that can form the basis of impression analyses. The large amounts of information that people share could mean these techniques may serve in trial recruitment. A recent study regarding cancer trials showed that clinical trials were offered to patients only 20% of the time, but of those 75% accepted. Further investigation identified that 32% said they would be very willing to participate in a clinical trial if asked. If, using these techniques, it is possible to identify candidates for inclusion then studies can be bought to subjects; the statistics suggest that there is a good probability of a successful match benefitting the patients.
DISTRIBUTED ETL
Simple transformations can be achieved using standard libraries with MapReduce. Examples include mapping a raw date string to an ISO8601 format or populating standard and raw variables given the datatype. Assuming that rows are independent, then it is possible to parallelize mapping processes across many workers. By minimizing the time required to generate the transformed dataset, the requirement for holding many copies of intermediate datasets can be mitigated as it becomes possible to regenerate the required datasets from the source data on demand (or even continuously). The MapReduce approach can assist ETL processes; mapper nodes can be configured to execute SQL queries against a database making it possible to parallelize extracting data from databases. An open source tool called Apache Sqoop uses this technique to extract data from a RDBMS into HDFS. If an enterprise service bus or equivalent messaging layer is attached as a Spout, then real time transformations are possible feeding data directly into standardized analysis platforms.
STUDY STATUS REPORTS
Much of the data in clinical studies needs to be processed to provide overall metrics for the study; for example, how many CRFs pages need reviewing? These status metrics need to be processed from the point at which changes are made (the CRF level) up to the subject, site or study level. The status at any level is constituted of the status of its children (and their children, etc.). Viewing the hierarchy as a tree, it is possible to split the overall calculations as a series of sub-calculations based on bifurcation points, which can be processed independently. The child calculations are recursively computed upwards, with the result of each level giving the status of its parent. Such hierarchical review is possible with MaxisIT’s MaxisIT’s Analytics & Reporting platform which offers features like drag-n-drop analytical data modelling, statistical as well as data-driven matrices-based analytics configurator, reports portfolio manager, multiple visualizations and reporting structure support, template management, role-based access controls, version controls, interactive dashboard, and analytical sandbox environment with automated refresh – configurability, reusability and usability.
CONCLUSION
Big Data is a valuable approach and as an industry we should have a strategy for incorporating it into our data practices. The advantages that can be gained by enabling rapid processing of large amounts of data and being able to use the results to make informed decisions are significant. As we see with the Apache Hadoop project, many tools are built up around the central platform that lower the cost of adoption for organizations seeking to embrace these new technologies. The definition of Big Data being a problem that necessitates continual innovation presents many opportunities for improvements in what can be achieved with data generated by clinical studies, benefitting the industry and those depending on what it produces. Data analysis becomes less bound by data logistics; both in terms of storage considerations and time-to-generate considerations; this will open the organization to wider ranges of approaches to data processing and the types of analytics that can be attempted. The process becomes data driven, which is where it should be – subjects have contributed their data and we should be able to get maximum value from this for their sake.
MaxisIT
At MaxisIT, we clearly understand strategic priorities within clinical R&D, and we can resonate that well with our similar experiences of implementing solutions for improving Clinical Development Portfolio via an integrated platform-based approach; which delivers timely access to study specific as well as standardized and aggregated clinical trial operations as well as patient data, allows efficient trial oversight via remote monitoring, statistically assessed controls, data quality management, clinical reviews, and statistical computing. Moreover, it provides capabilities for planned vs. actual trending, optimization, as well as for fraud detection and risk-based monitoring. MaxisIT’s Integrated Technology Platform is a purpose-built solution, which helps Pharmaceutical & Life sciences industry by “Empowering Business Stakeholders with Integrated Computing, and Self-service Dashboards in the strategically externalized enterprise environment with major focus on the core clinical operations data as well as clinical information assets; which allows improved control over externalized, CROs and partners driven, clinical ecosystem; and enable in-time decision support, continuous monitoring over regulatory compliance, and greater operational efficiency at a measurable rate”.
Clinical study designs are becoming increasingly complex. A growing number of studies are using adaptive designs and require decisions during the conduct of the study. At the same time there’s a growing demand for more amount of data, a larger variety of data types and time pressures on decision making. During clinical study conduct, scientists are under high time pressure as they need to manage a multitude of tasks, such as medical data review, signal detection on clinical study and project level, conducting preliminary analysis, preparing for database closure and working on publications and presentations.
Above all, clinical scientists are expected to drive innovation in pharmaceutical drug development with new clinical study designs, new assays and new ways to look at data. Innovative thinking requires time and a mind at ease, a contradicting requirement in the busy world of clinical studies. Since the evaluation of data is very often done with sub-optimal tools and processes requiring a lot of manual work, the clinical scientists have neither the time not space needed to turn creative and develop new ideas.
An effective and streamlined data flow from data capture to decision making can support the scientists in their intrinsic key responsibility to innovate drug development. The specific deliverables of an improved data flow must focus on two aspects:
Such improvements need to be achieved on the back of a high economic pressures for further improved operational efficiency and continuously high levels of data quality and regulatory compliance.
RETHINKING THE FLOW OF DATA
Addressing the scientists’ needs according to these requirements is a tall order. It requires a comprehensive approach looking at the systems, data standards and business processes in a combined fashion. Standardization is the underlying common characteristics to all of these because it offers re-usability and reduced time and effort.
Specifically, there are 3 topics that require consideration:
Finally, all functions involved need to be absolutely clear on their contribution and responsibilities across the entire data flow. In addition, there needs to be a clear distinction between mandatory process steps and deliverables versus areas where flexibility is possible and welcome.
SIMPLIFYING THE DATA FLOW AND TOOLS FOR CLINICAL STUDIES
The key design principles for the future system landscape was to minimize the number of tools and databases, eliminate redundant data storage where possible, and use the same tools or platforms across functions. The different options for the data flows needs to be reduced to one preferred way of working: on-line EDC data capture and access to clinical data via a graphical data review tool.
Tools and Platforms:
Data flow:
Data Standards
PROVIDING SPEEDY ACCESS TO STUDY DATA
A key requirement for clinical scientists is early and speedy access to study data. This can be greatly supported by the use of global data standards. A Gartner report showed that CDISC data standards can reduce the time for study setup by up to 80% and the time for data extraction into a usable format by up to 50%. Such time savings translate directly into the thinking time and space for scientists for decision making.
The redesigned data flow offers a variety of components for early and speedy access to study data.
STANDARDIZING DATA FORMATS AND DISPLAYS
Data standardization supports the fast database setup and enables a fast data flow during study conduct. Beyond that, standards are extremely valuable when it comes to integrated analysis reaching across studies. Finally, standards are a strong enabler for presenting data in an interpretable fashion. Downstream tools need to find variable names and types based on standardized names, and scientists are becoming used to this nomenclature.
For the re-designed data flow, CDISC data standards play a key role:
NEW RESPONSIBILITIES FOR CLINICAL SCIENCE
Early and speedy access to clinical data during study conduct is a privilege which comes with responsibilities.
In order to work with data, the clinical scientists need to acquaint themselves with the concept of data models. As a prerequisite to data exploration, the meaning and interpretation of the variables in data sets need to be understood.
When receiving data early during study conduct, it needs to be understood that the data are not clean. This should not cause friction in a team but should be understood by all parties involved.
Clinical scientists need to apply the concept of data exploration: first comes a question, then the data are explored using an adequate tool to get an answer to the question. Following the format of question and answer should help to look at data in a structured manner, without getting lost in a jungle of data.
CONCLUSION
The key elements to enable scientific innovation in drug development are
The daily transactions in drug development, however, frequently do not provide room for both, data availability and thinking time. Correspondingly, an improved data flow facilitated by an integrated data management platform coupled with data visualization tools can encourage innovation while maintaining overall efficiency and regulatory compliance.
About MaxisIT
At MaxisIT, we clearly understand strategic priorities within clinical R&D, and we can resonate that well with our similar experiences of implementing solutions for improving Clinical Development Portfolio via an integrated platform-based approach; which delivers timely access to study specific as well as standardized and aggregated clinical trial operations as well as patient data, allows efficient trial oversight via remote monitoring, statistically assessed controls, data quality management, clinical reviews, and statistical computing. Moreover, it provides capabilities for planned vs. actual trending, optimization, as well as for fraud detection and risk-based monitoring. MaxisIT’s Integrated Technology Platform is a purpose-built solution, which helps Pharmaceutical & Life sciences industry by “Empowering Business Stakeholders with Integrated Computing, and Self-service Dashboards in the strategically externalized enterprise environment with major focus on the core clinical operations data as well as clinical information assets; which allows improved control over externalized, CROs and partners driven, clinical ecosystem; and enable in-time decision support, continuous monitoring over regulatory compliance, and greater operational efficiency at a measurable rate”.
SDV is a very expensive process due to the time required to go through all the data at the various investigator sites. However, if we can target the patients and specify items the CRAs should look at when they visit a site, then the CRAs can spend more time looking at the important data but still spend less time overall. Although this may not be too useful for small studies, for large and mega-trials this can and has already saved millions. This approach is also being encouraged by the FDA and EMA as a possible method for improving the quality of trials and trial data. The key aspect here is defining the risk associated with a site, and how to change this over time.
CURRENT ISSUES WITH SDV
Quality risk management in clinical trials is often interpreted as risk elimination when it comes to SDV.
Pharmaceutical companies attempt to eliminate risk by performing 100% SDV. However, the cost of on-site monitoring is now around a third of the cost of a trial, so performing 100% SDV is a very expensive method of eliminating risk. Also, on-site monitoring involves many more tasks than just SDV. Tasks that are important for the overall quality and compliance, including GCP measures must also be performed.
The challenge for the monitor is then to perform 100% SDV and all the other tasks without spending a considerable number of hours at the investigator site. This leads to rushing the site visit, and thereby either missing issues, especially if they are related to cross checks between different questions on multiple pages, or not having enough time to clarify and explain how to avoid certain issues
RISK OF NOT PERFORMING 100% SDV
Studies have shown that only a very small percentage of data is changed due to 100% SDV, and the effect of this change on the primary analysis is negligible. Many of the data issues are identified and queried by Data
Management during screening rules checks and consistency checks. Therefore quality risk is not directly affected by 100% SDV. It was often thought that regulators preferred 100% SDV, and therefore not to do this may raise quality risk concerns.
However, papers from FDA (Guidance for Industry – Oversight of Clinical Investigations – A Risk-Based Approach to Monitoring, August 2011) and EMA (Reflection Paper on Risk-Based Quality Management in Clinical Trials, August 2011) have positively encouraged pharmaceutical companies to abandon the 100% SDV approach in preference to a more risk-based approach. So the risk of problems from regulators for following such an approach is now also no longer an issue.
WHAT IS A CENTRALIZED MONITORING RISK-BASED SDV APPROACH
Centralized monitoring can be thought of as moving a little bit away from the manual and subjective process to an automated and logical process. It is moving away from having humans look at and check the data to a process where all the data is checked by programs. As checks are then programmed centrally and run on all patient data, it then becomes possible to identify key data issues for the monitor to check, and therefore in effect ask the monitor to target their SDV and mostly check the data with issues.
This process has a risk associated with it, as the monitor is not performing all the checks manually, and they are not reviewing 100% of the data. However, the advantages of automating these processes far outweigh the benefits of the previous manual process. The process now becomes something like this:
1) Assign risk
Receive data from investigator sites
Receive feedback from CRAs.
Use programmed centralized checks to verify the data for consistency and accuracy as well as fraud.
Risk is assigned to each site based on the data checks, CRA feedback and knowledge from previous and ongoing collaboration.
What data of which patient the CRAs should check is identified based on the risk.
2) Data queries
Investigator sites receive queries raised by automated checks.
Submit centralized check programs regularly to monitor data quality of the latest data as an ongoing process.
3) Monitoring visits
If a site has some types of data issues coming up consistently, then inform the CRA to provide more training to reduce those issues
Inform CRAs about which patients to check and to which level for each site they are responsible for.
CRAs only check the data they are instructed to check.
CRAs then have more time to check:
ADVANTAGES OF A CENTRALISED RISK-BASED SDV APPROACH
The advantage of a centralized risk-based SDV approach is that systemic errors are easy to identify by looking at data trends and protocol violators. This means that if a site has misunderstood something this will become obvious.
It also means that all sites are being checked regularly by the automated checks on the latest data, and there is no need to wait until a monitoring visit is performed.
Automated programs also have the advantage that data errors, outliers, missing and inconsistent data are identified with logic rather than the luck of the eye, and more complex fraud checks and statistical analysis can be programmed very easily. Site characteristics and performance metrics can also be monitored over time by looking at high screening failure rates, eligibility violations and delays in reporting the data.
All this means that the CRAs have fewer data to review when they are at the site, which leaves them with time to both verify the source data and check the data to ensure it makes sense. They also have extra time to do more GCP and Process checks at the site, provide more training if required and so on. CRAs will then be able to visit sites with issues more often and spend a long time there and visits sites without issues less frequently. This all helps to improve the quality of the trial and the data.
This approach will not only increase the chances of identifying data issues, both random and systematic, it will also help to check for fraud and increase the quality of the trial. As more time will be spent on automatic checks, and less on on-site monitoring, the overall cost of on-site monitoring will be reduced. This saving will increase as the size of the trial increases from small to medium to mega-trials.
Today, cloud-based integrated platforms can assimilate source data and provide one source of truth for CRA’s to perform automated checks. With real-time visual analytics, these platforms make it easy to verify source data more effectively leading to improved site monitoring, better compliance and faster reporting of clinical data.
About MaxisIT
At MaxisIT, we clearly understand strategic priorities within clinical R&D, and we can resonate that well with our similar experiences of implementing solutions for improving Clinical Development Portfolio via an integrated platform-based approach; which delivers timely access to study specific as well as standardized and aggregated clinical trial operations as well as patient data, allows efficient trial oversight via remote monitoring, statistically assessed controls, data quality management, clinical reviews, and statistical computing. Moreover, it provides capabilities for planned vs. actual trending, optimization, as well as for fraud detection and risk-based monitoring. MaxisIT’s Integrated Technology Platform is a purpose-built solution, which helps Pharmaceutical & Life sciences industry by “Empowering Business Stakeholders with Integrated Computing, and Self-service Dashboards in the strategically externalized enterprise environment with major focus on the core clinical operations data as well as clinical information assets; which allows improved control over externalized, CROs and partners driven, clinical ecosystem; and enable in-time decision support, continuous monitoring over regulatory compliance, and greater operational efficiency at a measurable rate”.