-
WHAT WE DO
- CLINICAL SOLUTIONS
- SERVICES
-
WHO WE ARE
-
RESOURCES

SDV is a very expensive process due to the time required to go through all the data at the various investigator sites. However, if we can target the patients and specify items the CRAs should look at when they visit a site, then the CRAs can spend more time looking at the important data but still spend less time overall. Although this may not be too useful for small studies, for large and mega-trials this can and has already saved millions. This approach is also being encouraged by the FDA and EMA as a possible method for improving the quality of trials and trial data. The key aspect here is defining the risk associated with a site, and how to change this over time.
CURRENT ISSUES WITH SDV
Quality risk management in clinical trials is often interpreted as risk elimination when it comes to SDV.
Pharmaceutical companies attempt to eliminate risk by performing 100% SDV. However, the cost of on-site monitoring is now around a third of the cost of a trial, so performing 100% SDV is a very expensive method of eliminating risk. Also, on-site monitoring involves many more tasks than just SDV. Tasks that are important for the overall quality and compliance, including GCP measures must also be performed.
The challenge for the monitor is then to perform 100% SDV and all the other tasks without spending a considerable number of hours at the investigator site. This leads to rushing the site visit, and thereby either missing issues, especially if they are related to cross checks between different questions on multiple pages, or not having enough time to clarify and explain how to avoid certain issues
RISK OF NOT PERFORMING 100% SDV
Studies have shown that only a very small percentage of data is changed due to 100% SDV, and the effect of this change on the primary analysis is negligible. Many of the data issues are identified and queried by Data
Management during screening rules checks and consistency checks. Therefore quality risk is not directly affected by 100% SDV. It was often thought that regulators preferred 100% SDV, and therefore not to do this may raise quality risk concerns.
However, papers from FDA (Guidance for Industry – Oversight of Clinical Investigations – A Risk-Based Approach to Monitoring, August 2011) and EMA (Reflection Paper on Risk-Based Quality Management in Clinical Trials, August 2011) have positively encouraged pharmaceutical companies to abandon the 100% SDV approach in preference to a more risk-based approach. So the risk of problems from regulators for following such an approach is now also no longer an issue.
WHAT IS A CENTRALIZED MONITORING RISK-BASED SDV APPROACH
Centralized monitoring can be thought of as moving a little bit away from the manual and subjective process to an automated and logical process. It is moving away from having humans look at and check the data to a process where all the data is checked by programs. As checks are then programmed centrally and run on all patient data, it then becomes possible to identify key data issues for the monitor to check, and therefore in effect ask the monitor to target their SDV and mostly check the data with issues.
This process has a risk associated with it, as the monitor is not performing all the checks manually, and they are not reviewing 100% of the data. However, the advantages of automating these processes far outweigh the benefits of the previous manual process. The process now becomes something like this:
1) Assign risk
Receive data from investigator sites
Receive feedback from CRAs.
Use programmed centralized checks to verify the data for consistency and accuracy as well as fraud.
Risk is assigned to each site based on the data checks, CRA feedback and knowledge from previous and ongoing collaboration.
What data of which patient the CRAs should check is identified based on the risk.
2) Data queries
Investigator sites receive queries raised by automated checks.
Submit centralized check programs regularly to monitor data quality of the latest data as an ongoing process.
3) Monitoring visits
If a site has some types of data issues coming up consistently, then inform the CRA to provide more training to reduce those issues
Inform CRAs about which patients to check and to which level for each site they are responsible for.
CRAs only check the data they are instructed to check.
CRAs then have more time to check:
ADVANTAGES OF A CENTRALISED RISK-BASED SDV APPROACH
The advantage of a centralized risk-based SDV approach is that systemic errors are easy to identify by looking at data trends and protocol violators. This means that if a site has misunderstood something this will become obvious.
It also means that all sites are being checked regularly by the automated checks on the latest data, and there is no need to wait until a monitoring visit is performed.
Automated programs also have the advantage that data errors, outliers, missing and inconsistent data are identified with logic rather than the luck of the eye, and more complex fraud checks and statistical analysis can be programmed very easily. Site characteristics and performance metrics can also be monitored over time by looking at high screening failure rates, eligibility violations and delays in reporting the data.
All this means that the CRAs have fewer data to review when they are at the site, which leaves them with time to both verify the source data and check the data to ensure it makes sense. They also have extra time to do more GCP and Process checks at the site, provide more training if required and so on. CRAs will then be able to visit sites with issues more often and spend a long time there and visits sites without issues less frequently. This all helps to improve the quality of the trial and the data.
This approach will not only increase the chances of identifying data issues, both random and systematic, it will also help to check for fraud and increase the quality of the trial. As more time will be spent on automatic checks, and less on on-site monitoring, the overall cost of on-site monitoring will be reduced. This saving will increase as the size of the trial increases from small to medium to mega-trials.
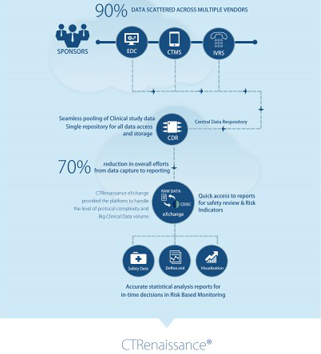
Today, cloud-based integrated platforms can assimilate source data and provide one source of truth for CRA’s to perform automated checks. With real-time visual analytics, these platforms make it easy to verify source data more effectively leading to improved site monitoring, better compliance and faster reporting of clinical data.
About MaxisIT
At MaxisIT, we clearly understand strategic priorities within clinical R&D, and we can resonate that well with our similar experiences of implementing solutions for improving Clinical Development Portfolio via an integrated platform-based approach; which delivers timely access to study specific as well as standardized and aggregated clinical trial operations as well as patient data, allows efficient trial oversight via remote monitoring, statistically assessed controls, data quality management, clinical reviews, and statistical computing. Moreover, it provides capabilities for planned vs. actual trending, optimization, as well as for fraud detection and risk-based monitoring. MaxisIT’s Integrated Technology Platform is a purpose-built solution, which helps Pharmaceutical & Life sciences industry by “Empowering Business Stakeholders with Integrated Computing, and Self-service Dashboards in the strategically externalized enterprise environment with major focus on the core clinical operations data as well as clinical information assets; which allows improved control over externalized, CROs and partners driven, clinical ecosystem; and enable in-time decision support, continuous monitoring over regulatory compliance, and greater operational efficiency at a measurable rate”.
21 Jul 2020

6 Jun 2018